Fucosidosis
Fucosidosis
Fucosidosis is caused by the lack of the enzyme alpha-fucosidase.
There are two different types of fucosidosis. They are characterized by the age of onset and type of physical and mental manifestations.
In the past, researchers described two types of this condition based on symptoms and age of onset, but current opinion is that the two types are actually a single disorder with signs and symptoms that range in severity.
How Common is it?
Fucosidosis is a very rare lysosomal storage disease and though the incidence worldwide is not known with any certainty, it is estimated to affect less than 1:2,000,000.

Type I & Type II
Type I usually presents in the first 3–18 months of life with features typical of lysosomal storage diseases including coarsening of facial features, large liver, spleen and/or heart and dysostosis multiplex (abnormal bone formation that is found in multiple bones of the body) seen on x-ray. Cherry red spots may be present on ophthalmology evaluation. Cherry red spots are spots on the retina that have storage of sugar chains, causing the remainder of the healthy retina to look brighter. Mental retardation and seizures are also present. Additionally, sweat chloride may be elevated.
Type II fucosidosis presents between 12 and 24 months of life. Individuals usually have mild coarsening of facial features, dysostosis multiplex, mental retardation enlarged liver, spleen and/or heart involvement. In addition, angiokeratomas (superficial blood vessel dilation over which wart like growths occur) may be present. Another feature of type II fucosidosis is the presence of twisted blood vessels within the membrane covering of the eyeball and inner eyelid.
Treatments:
Individuals with Fucosidosis should have routine follow-up with Genetics, Neurology, Ophthalmology, and other specialists as needed. Currently, there is no cure to stop the progression of symptoms of Fucosidosis and treatment is aimed at addressing the individual problems as they arise.
For some Glycoprotein diseases, including Fucosidosis, bone marrow transplant has been trialed as an experimental therapy but there are no conclusive results on the long term benefits. Ask your specialist for more information on this.
Patient Support Groups:
- ISMRD is the international support group for this disorder
- ISMRD’s Facebook page; this is our closed group for families only
- ISMRD’s public Facebook page for this disorder
- UK MPS Society is the support group for MPS and related disease in the United Kingdom
- Other MPS societies around the world may also offer some support
More Facts & Details:
What is Fucosidosis?
Fucosidosis is one of nine identified Glycoprotein Storage Diseases. These inherited diseases are part of a larger group of disorders called Lysosomal storage diseases. Lysosomes are membrane-bound compartments found in the cells of the body. These compartments contain enzymes, which are responsible for the breakdown of many different oligosaccharides (long sugar chains). These sugar chains are continuously made and broken down in our bodies, and this process is necessary for appropriate mental and physical development. Each enzyme in the lysosome is responsible for a certain step in the breakdown of the sugar chains.
When an enzyme is not working, it leads to the build up of the sugar chains in the lysosome. In Fucosidosis, the specific enzyme that is absent is called alpha- fucosidase. The build up of oligosaccharide sugars that is caused, is gradual and interferes with the correct function of the cell. This build up is gradual and eventually leads to the clinical features of Fucosidosis. Features may progress in severity over time.
In Fucosidosis, the specific enzyme that is absent is called alpha-fucosidase.
What are the clinical features of Fucosidosis?
There are two different types of Fucosidosis. They are characterized by the age of onset and type of physical and mental manifestations. Type I usually presents in the first 3–18 months of life with features typical of lysosomal storage diseases including coarsening of facial features, organomegaly (large liver, spleen and/or heart), and dysostosis multiplex (abnormal bone formation that is found in multiple bones of the body) seen on x-ray. Cherry red spots may be present on ophthalmology evaluation. Cherry red spots are spots on the retina that have storage of sugar chains, causing the remainder of the healthy retina to look brighter. Mental retardation and seizures are also present. Additionally, sweat chloride may be elevated.
Type II Fucosidosis presents between 12 and 24 months of life. Individuals usually have mild coarsening of facial features, dysostosis multiplex, mental retardation and organomegaly. In addition, angiokeratomas (superficial blood vessel dilation over which wartlike growths occur) may be present. Another feature of type II Fucosidosis is the presence of twisted blood vessels within the membrane covering of the eyeball and inner eyelid.
How is Fucosidosis inherited?
Fucosidosis is not contagious and cannot be “caught.” It is a genetic condition, which means that it is caused by a change in the instructions that direct the way our bodies grow and develop. These instructions are called genes. People have two copies of all their genes, including the gene for Fucosidosis (alpha- fucosidase). One copy is inherited from the mother in the egg, and one from the father in the sperm.
Only when there is a change in the gene code is there a possibility that the disease will occur. For a person to have Fucosidosis, they must inherit changes in both of their alpha-fucosidase genes resulting in instructions that do not function properly. This is known as autosomal recessive inheritance.
For a couple to have a child with Fucosidosis, both parents must have at least one changed copy of the alpha-fucosidase gene which they both pass on to their child. Parents do not have control over which genes they pass on to their children.
If a person has one changed copy of the alpha-fucosidase gene and one normal copy of the alpha-fucosidase gene they are said to be a “carrier” of the condition and will not show any symptoms of Fucosidosis. If two parents are both carriers, they have a 1 in 4 (25%) chance of having a child with Fucosidosis in each pregnancy.
What testing is available to determine if my child or I have Fucosidosis?
Testing for Glycoprotein storage diseases is typically performed in conjunction with a genetics evaluation. A genetics team takes into account the medical history and clinical features of a patient to determine what type of genetic testing is appropriate. For the diagnosis of an Glycoprotein storage disease, a urine test should show increased oligosaccharides. To determine if the patient has Fucosidosis, the urine test should be followed by a blood test or skin biopsy. The blood or skin sample should show decreased amounts of the enzyme alpha- fucosidase.
For families who have had a child diagnosed with Fucosidosis, prenatal diagnosis is available in future pregnancies by looking at alpha-fucosidase activity through Chorionic Villus Sampling (CVS) or amniocentesis. Prenatal diagnosis by detection of alpha-fucosidase gene changes is also available for families in which the responsible gene changes have been identified.
What type of treatment is available for Fucosidosis?
Individuals with Fucosidosis should have routine follow-up with Genetics, Neurology, Ophthalmology, and other specialists as needed. Currently, there is no cure to stop the progression of symptoms of Fucosidosis and treatment is aimed at addressing the individual problems as they arise.
For some Glycoprotein diseases, bone marrow transplant has been trialed as an experimental therapy but there are no conclusive results on the long term benefits. Ask your specialist for more information on this.




Medical and Research Information
- GeneTests: A list of labs testing for Fucosidosis.
- Genetic Home Reference: This has an excellent description of Fucosidosis with good references to other information and resources.
- OMIM: Technical information about the genetics of Fucosidosis. OMIM is a site developed for scientists and medical specialists and contains both general and highly technical information. Access to this site is free.
- Orphanet: A database dedicated to information on rare diseases and orphan drugs. Access is free of charge.
- Clinicial Trials. A registry of federally and privately supported clinical trials conducted in the United States and around the world. This site gives information about each trial’s purpose, who may participate and the location.
Meet People with Fucosidosis
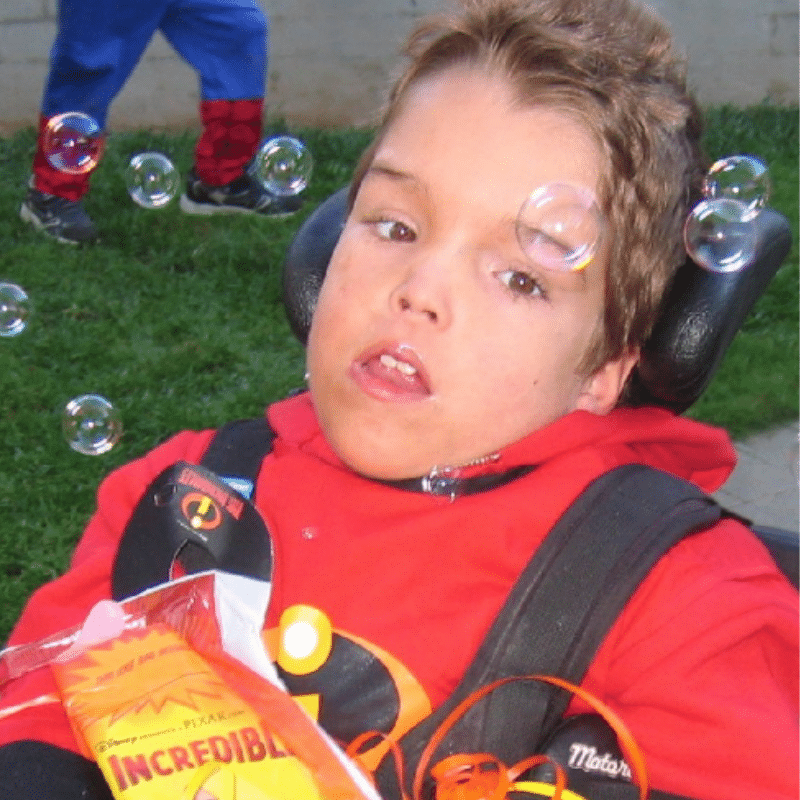
Matthew
Matthew was born a bright and happy boy, but his world was gradually clouded by a rare, degenerative disorder called Fucosidosis. This condition, caused by the absence of a vital enzyme, led to the accumulation of certain sugars in his body, slowly robbing him of his health.
As the disease progressed, Matthew’s once vibrant spirit was dimmed. He lost the ability to move and speak, and was plagued by frequent seizures. His frail body was further weakened by respiratory infections, eventually succumbing to a devastating bout of double pneumonia at the age of 10.
In 1998, when Matthew was first diagnosed, the internet was still in its infancy. Armed with only a few research articles, we felt utterly isolated. The weight of uncertainty and the fear of the unknown were overwhelming. Joining the International Society for Mannosidosis and Related Diseases (ISMRD) was a turning point. We found solace in connecting with other families facing similar challenges. The 2007 ISMRD conference in Michigan was a pivotal moment, where we met other Fucosidosis families and researchers working on potential treatments.
Today, his mom, Carolyn, is the President of ISMRD. She is proud to be part of a community that’s driving progress in rare disease research. The continuing development of an enzyme replacement therapy for Fucosidosis is a testament to the power of hope, perseverance, and collective effort.
Matthew’s legacy lives on, inspiring us to continue our fight for a future where rare diseases are no longer silent.

Nur
My name is Nur – I’m also known as Nunu or Noodlebug. I’m funny, cheeky and sassy and a total chatterbox.
I have an ultra rare title: I have two major diagnoses, one of severe Juvenile Idiopathic Arthritis and the other of Fucosidosis.
I had a double hip replacement when I was fourteen and I’m in a wheelchair, but I can still walk short distances although my Fucosidosis messes with my balance. I started college this year and I’m loving it.
In the past few years I had endured major operations, countless injections on a weekly basis, chemo, and these days a visit every month onto the wards at UCLH for infusions. I treat these appointments as social gatherings and hold court on the infusion suite.
In two weeks time I will once again prove that I’m a miracle, as I will surpass my predicted life expectation – in good health – as I will turn twenty years old.
Stephen & Lauren: We are siblings both with the ultra rare condition of Fucosidosis, but aren’t affected exactly the same.

Stephen
I’m 29 and struggle with joint stiffness, so I walk and move slowly in a very laboured way.
I don’t have a lot of stamina and get frequent colds and stomach upsets. The colds lay me low for quite a while, I can however do very limited occasional work at our local mail centre.
I enjoy watching sports but don’t play any.

Lauren
I’m 24 and I have much more stamina, recovering from minor illness very quickly.
I’m mentally much younger so I’m not allowed out on my own, I would be too vulnerable.
I have quite a lot of red marks, angiokeratomas on my body but they do not bother me.
I enjoy music and dancing