Glycoprotein Diseases
What are Glycoprotein Diseases?
Glycoprotein and Related Storage Diseases are very rare, progressive, largely untreatable metabolic enzymatic defects inherited by children from both parents. The worldwide incidence for these diseases as a group has not yet been determined accurately, though they can certainly be classified as ultra-rare. The course of these disorders affects multiple systems of the body and clinical symptoms may vary patient-to-patient, and even among siblings. For most children the implications are eventual loss of mental and physical functions, and a premature death.
As the name implies, glycoproteins are complex compounds composed of a protein and a carbohydrate. There are many different types of glycoproteins and they are found in abundance in all kinds of cells, including those of the brain. Glycoproteins play many roles in cells; some of these are well known, but others have yet to be discovered. Known roles include acting as an agent for communication between cells and assisting in maintaining the cell’s structure. The carbohydrate portion of glycoproteins is usually made of combinations of sugar molecules such as glucose, galactose, mannose and fucose, which are collectively known as oligosaccharides.
Normal functioning of the cell is characterized by the continual degradation of glycoproteins by enzymes within the lysosomes, which are membrane-bound compartments in the cell and essentially the cell’s recycling center. Specific enzymes are needed along each “step” of the recycling process in order that the continual, complete breakdown of these chains of glycoproteins is carried out. Any malfunction along the way results in the premature termination of the process with a resulting accumulation of the undegraded material within the lysosome.
Patterns of Inheritance - How do children get Glycoprotein Storage diseases?
Glycoprotein Storage Diseases are autosomal recessive conditions, meaning that a defective trait or mutation passed on from both parents is required for the disease to be expressed. Every person carries two copies of a gene, one of which they pass on to their offspring (the other provided by the second parent). In many cases, we all have a mutation or defect in some of our genes, but do not show signs of the defect because the other gene is able to allow us to function “normally.” In autosomal recessive diseases, the condition occurs when both copies of a gene are defective.
Since each parent donates a single gene copy to each offspring, there are four possible inheritance patterns for diseases to occur.
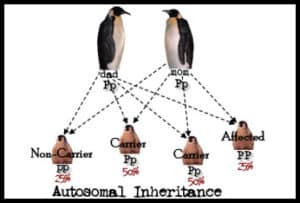
Since most people can live apparently normal lives with one healthy half of a gene pair, recessive gene disorders are relatively rare. However, as shown above, 50% of people inheriting an unexpressed, or recessive, gene defect are known as carriers. Carriers have the ability to continue a family’s mutation to the next generation. Over time, though, the mutation will become diluted unless a carrier mates with another carrier.
Two carrier parents have a 25% chance of giving birth to a child with an autosomal recessive disease. Because these odds occur for each pregnancy, it is possible, though not likely, that a family may have more than one child with a genetic disease. There is a greater likelihood, 50%, that offspring of two such parents will be a carrier and continue to pass on the defect to future generations. Lastly, a 25% chance exists that a child born to two carrier parents will be completely healthy. Such a child inherits two good gene copies and, thus, will not pass the gene defect to its heirs.
For couples who have a known history of the same recessive genetic disease in their families, carrier testing is a possibility to assist with family planning. However, testing for a genetic defect without such prior knowledge or history is not possible or recommended for most autosomal recessive diseases.
Storage in Lysosomal Diseases: Why does storage cause disease?
[A comic created for ISMRD by noted cartoonist and illustrator, Denis Rodier]

In the Lysosomes, or Recycling Center of the Cell, our penguins work hard and efficiently to move large bags of undegraded compounds, or substrates (denoted by the “S” on each bag). Each bag is broken down and sent back to the body to be reused.

In some people, our penguins can’t do their job efficiently because of a genetic defect that prevents the bags of waste from moving through the Recycling Center. Most commonly, this defect is found in the enzyme whose job it is to bust up certain bags of substrates. The result is a slowdown in the workflow, resulting in substrates not moving through the Lysosome.

Over time work in the Recycling Center comes to a virtual standstill and the bags of substrates begin to build up in the Lysosomes. When this happens the person affected displays a multitude of symptoms that cause pain, neurologicaldamage and a shortened lifespan.




The Nine Types of Glycoprotein Storage Diseases
There are nine different types of Glycoprotein Storage Diseases. Seven of the diseases are caused by a defect in glycoprotein degradation, while two others are characterized by trafficking errors that limit normal targeting of digestive enzymes, which are themselves glycoproteins, to the lysosomes and prevent the lysosome from functioning normally.
Find our more about each of the nine types of Glycoprotein Storage Diseases, and connect with Facebook Groups for each Disease, by clicking on the link to the Disease Page from the main menu or the category list on this page.
Caused by a defect in glycoprotein degradation:
- Alpha-Mannosidosis
- Aspartylglucosaminuria
- Beta-Mannosidosis
- Fucosidosis
- Galactosialidosis
- Schindler Disease
- Sialidosis, also known as Mucolipidosis I
Caused by tracking errors that limit normal targeting of digestive enzymes:
- Mucolipidosis alpha/beta, encompassing I-Cell Disease (Mucolipidosis II), Mucolipidosis II/III and Pseudo-Hurler Polydystrophy (Mucolipidosis III)
- Mucolipidosis III Gamma is a variant of Mucolipidosis III alpha/beta