

Sarah and her family’s journey with AlphaMannosisosis is presented in an educative and positive light.
Sarah’s story about Living with Alpha-Mannosidosis

This is Taryn’s Story about Living with Alpha-Mannosidosis and how the family of a teenager with a rare genetic disease copes with the affects of the disorder and how she gets her wish to meet her idols.

Our son and daughter, Timothy and Hollie are twins, born 14 November 1974. They were premature (about one month). There were some early indicators that it was more than just a rough start for them, but nothing really clear till they were about one year old and a number of milestones were not met on time.
This is their story told by their father John Forman

This is the ongoing journal of Robert Stark’s fight against Alpha-Mannosidosis via Bone Marrow Transplant. Families and caregivers of children with mannosidosis and other Lysosomal Diseases face complex decisions about the future. It is the hope of Robert’s parents, Kathleen and Mark, that the pages within will prove helpful when such decisions must be made.
This is Robert’s Road to Recovery after diagnosis and Bone Marrow Transplant

Luke was diagnosed with alpha-mannosidosis when he was 5. He underwent a bone marrow transplant in September 2010, 5 months after being diagnosed. We believe the bone marrow transplant has helped treat some of the symptoms of this disorder.
This is Luke’s Blog written by his family as he underwent Bone Marrow Transplant.

Saffy’s blog written by her parents documenting her journey through Bone Marrow Transplant.

Ryleigh’s parents, Kyle and Whitney have provided a website so that they can provide information on Ryleigh’s rare genetic disorder, Alpha-Mannosidosis. Ryleigh also has a Facebook page. Follow her journey as she goes through Bone Marrow Transplant.

It took us 16 years to learn our children’s correct diagnosis. Observing mine and other kids throughout many years, I came to believe that my sons’ delays were not all I’ve been told; there was something else standing between my kids and their ability to become what they want to be.
 My little “Ivan The Great” was diagnosed January 2011 with Galactosialidosis and am told it is EXTREMELY rare. I will never forget the day I found out his diagnosis and on top of that, that there is no cure or treatment for it. I felt like my heart was ripped right out from my chest. The whole world and how I perceived it changed instantly. I viewed the world and people in a whole new way.
My little “Ivan The Great” was diagnosed January 2011 with Galactosialidosis and am told it is EXTREMELY rare. I will never forget the day I found out his diagnosis and on top of that, that there is no cure or treatment for it. I felt like my heart was ripped right out from my chest. The whole world and how I perceived it changed instantly. I viewed the world and people in a whole new way.
 Madison’s mum April has put together a video in tribute to her beautiful daughter and is thrilled to be able to share it with other families.
Madison’s mum April has put together a video in tribute to her beautiful daughter and is thrilled to be able to share it with other families.
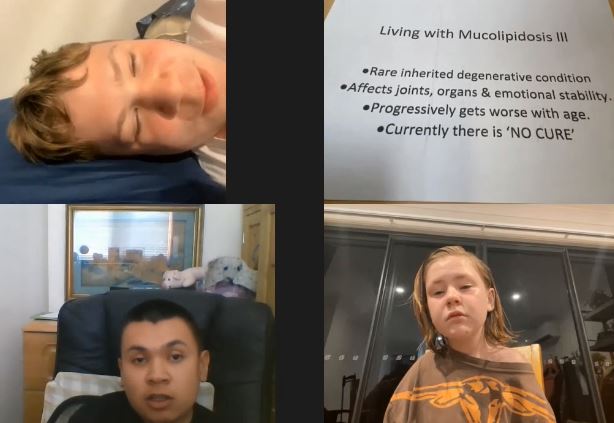
Meet three young ML III patients (Sam, Jesse-Rose and Damian) and their thoughts on how they live, what health problems they have, fears, desires… It is very unlikely that their story and thinking will not touch you.
Rare Disease Day 2022 – Living With ML III

Jenny Noble, from Tauranga New Zealand recounts their family’s experience with Mucolipidosis III alpha/beta, and details the dramatic improvements to Hayden and Sarah’s health and quality of life, following a trial of Pamidronate to treat the secondary bone disease that became such a significant problem in their teenage years.
Read about Hayden and Sarah’s journey with Mucolipidosis

Juanita talks about Damian and Jesse-Rose’s journey to diagnosis and beyond. She also says, If anyone asks me emotionally how we have been going, I let them know we are positive and doing what we can to make the kids lives easier. It’s hard to say out loud that there are times when I feel devastated, times when I feel like I could fall into a dark pit and never come out again. There are many times when I feel powerless in changing anything, so I pray! I pray that we find at the very least a treatment that means they have minimal pain, that they can have functioning useful lives and contribute to society. I pray mostly that we find a cure.
Whatever our journey is, it will be one that is perhaps more heartfelt and precious than it might have been prior to receiving this diagnosis.

A Family’s fight against Sialidosis
Sialidosis is one of the Oligosaccharide family of Lysosomal Storage Diseases. The International Society for Mannosidosis & Related Diseases is proud to present the story of Alexander Skojec and his family’s fight, through the intervention of his father, to raise awareness for this very rare disorder.

Tyler Huneault on national news
Tyler talks to channel A about his annual Garage Sale.

Hello, my name is Trajano Cerna. I am from Quito-Ecuador, and I live in the United States. I went to School of Medicine for 2 years in Ecuador. I enjoy walking and doing exercises in the park. And I like drinking natural juices.
My story starts here – according to my mother, I am a prematurely 7 month baby with a normal birth; but “to grow healthy” I was put in an incubator. My childhood was difficult, I had several medical problems due to falls and head bumps. The first serious medical issue related to my health was the low vision. I was diagnosed with severe myopia and astigmatism at 5 years old. When I was 16 years old, I went through my first epileptic crisis. That time I used to live in Ecuador. The falls and head bumps were more frequent during my adolescence. Due to this concern I decided to visit a neurologist doctor in Ecuador. His diagnosis was an issue related to the central nervous system, prescribing me the medication “Valpakine”. I took this medication for several years in Ecuador. In 2001, when I was 24 years old, I migrated to the United States. My first medical appointment in the United States was with a general doctor who concluded that I had chronic epilepsy. A new medication showed up to my daily life, called “Depakote”. I took this medication for about twelve years.
At that time, the excessive medications and the lack of information of my medical condition contributed to my health getting worse. And I got the following diseases: gastritis, ulcers, arthritis, osteosclerosis, low hemoglobin, and very low vision. Life seemed like it was coming at me. This situation encouraged me to look for help and I visited several doctors (neurologist, ophthalmologist, hematologist, epileptologist, and genetics). When I was 36 years old, I visited an epileptologist for the first time. He discovered the type of epilepsy that I have, called “Progressive Myoclonic Epilepsy”. One year later, I discovered other symptoms in my health: fatigue, anxiety, cherry red spots on retina, severe seizures, jerky body movements and difficulties to speak among others. With all these manifestations and millions of tests, the doctors concluded that my real disease is called “SIALIDOSIS”. This is a weird and unknown disease around the world.
From 2018 to 2020, I belonged to the National Institute of Health (NIH) in Maryland, United States. At NIH, I was part of an investigation team to support trial studies and scientific experiments. The goal was to collect information about this unknown disease.
In July of 2019, I attended an ISMRD conference in Atlanta, GA, called Supporting Families with Glycoprotein Storage Diseases. I met thoughtful people in there, all with the same goal, looking for help. At the moment there is no cure or treatment for Sialidosis. We are waiting for scientists, pharmacists or health institutions to be interested in helping us.
Nowadays, I am 47 years old, I live in New Jersey with the care of my parents and always willing to support the science and help other families with similar health conditions than mine.